")




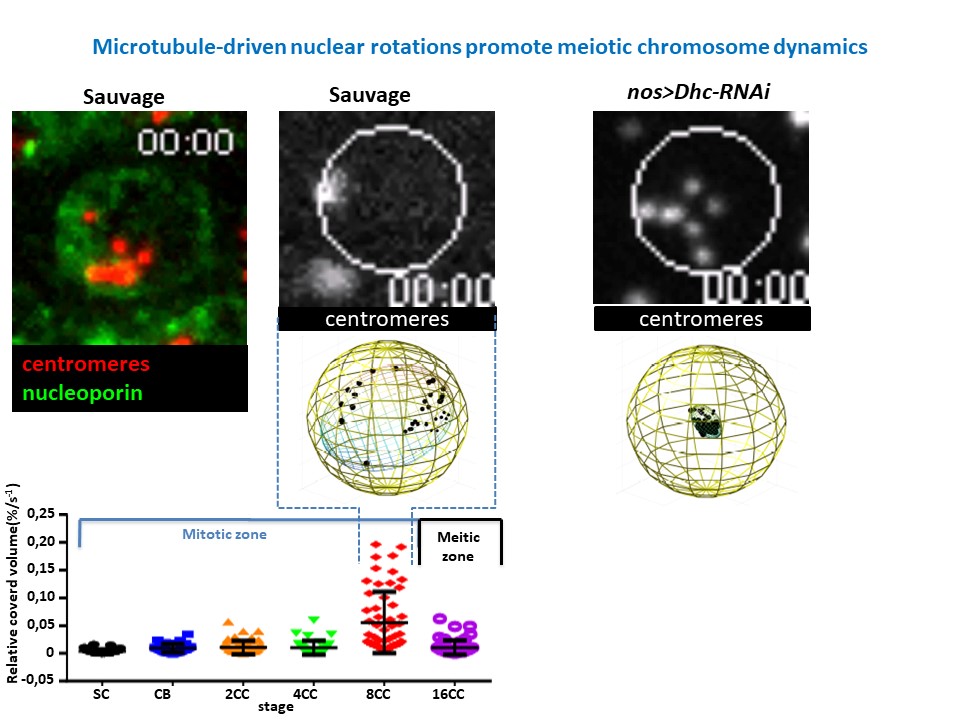
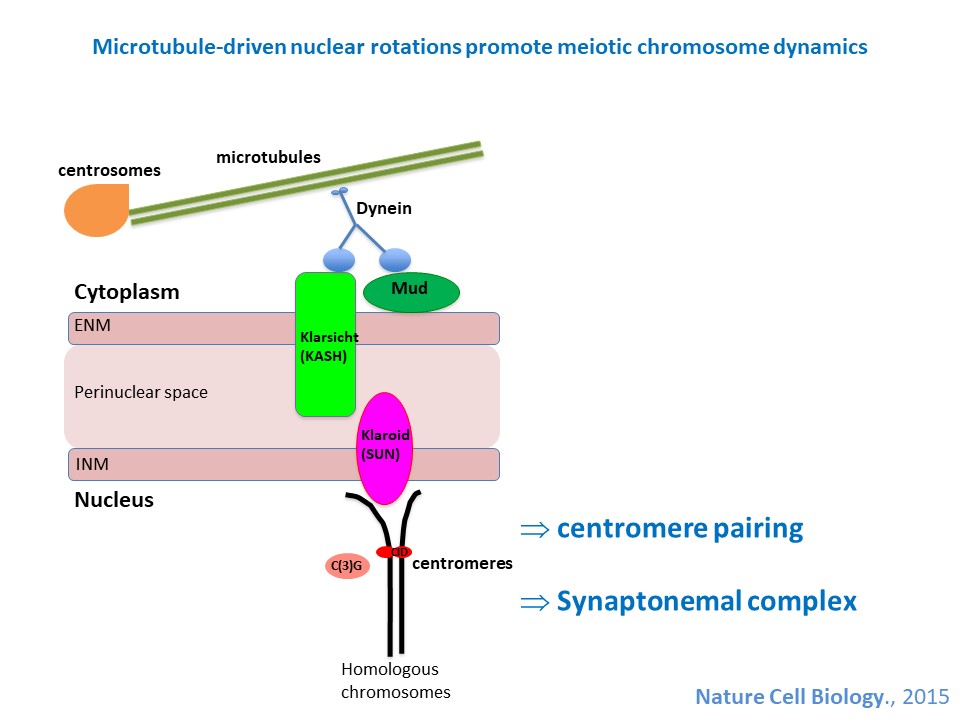
Developmental biology aims to study how cells divide and grow to form organs and entire organisms. Germ cells give rise to highly specialized cells called gametes used during sexual reproduction. During their differentiation, germ cells acquire all the genetic, cytoplasmic and epigenetic information to be transmitted to the next generation. In most species, this information is set up during the very first stages of germ cell development. view more... Exploring the first stages of germ cell development in any species is a challenge. These precursor cells are either transient or rare or hidden in complex tissues. The Drosophila ovary is an excellent model system to take up this challenge. It is a stem cell-based system, which can produce egg chambers throughout the life of the adult female. All the early stages of development are thus present at any time and can be studied throughout adulthood. Two types of stem cells, germline (GSC) and somatic (FSC), are located in a tiny specialized structure called the germarium at the anterior of each ovary. view more... The germarium is an organized structure divided into four functional regions. GSCs reside in the most anterior part of the mitotic zone, also called region 1. GSCs divide asymmetrically to self-renew and to generate a precursor cell called a cystoblast. The cytoblast then undergoes exactly four rounds of mitosis to produce a germline cyst made of 16 cells. During these mitoses, cytokinesis is not completed and abscission, the last step of cytokinesis, is aborted. All 16 cells thus remain connected through ring canals and share the same cytoplasm. After the last mitosis, cysts enter region 2a, which is the meiotic zone, and all 16 cells begin meiosis. However, during differentiation in region 2, only one cell per cyst remains in meiosis, while the 15 others exit meiosis and endoreplicate their DNA. Each phase in the germarium, region 1, 2a/2b and 3, lasts about 24 hours. It thus takes approximately 3 days to go from stem cell to egg chamber in the germarium. 1) How is the duration of cytokinesis regulated in germ cells? view more... We identified mutations in Drosophila Aurora-B and Cyclin-B genes, which regulate complete abscission in germline stem cells and incomplete abscission in differentiating germ cells. 2-1) Differential regulation of abscission between germline stem cells and differentiating daughter cells. We have found that AuroraB and CyclinB localize at the midbody, and that CycB/Cdk-1 promotes abscission, while AuroraB/Survivin inhibits it. Both complexes have other functions during the cell cycle, which are mainly the control of entry into M phase and the metaphase-anaphase transition. At these two stages, the regulation of AuroraB and Cdk-1 is well-described and a list of known regulators is available 12 The core machinery that drives the eukaryotic cell cycle is very conserved across evolution, but has only been extensively studied in a few model systems (Morgan, 2007). During animal development, the canonical cell cycle is modulated and adapted in different cell types. For example, the rapid divisions of zebrafish or Drosophila embryos are devoid of Gap phases, while mouse giant trophoblast cells or many larval cells in Drosophila become polyploid by endoreplicating their DNA without any mitosis (M) phase (Budirahardja and Gonczy, 2009). How specific developmental programs alter different steps of the cell cycle remains to be understood in most cases. In this respect, the last stages of cell division, when daughter cells become separated, are probably the most diverse, but also the least explored. In sea urchin embryos, the timing of cytokinesis is shifted, and the completion of cell division only occurs during the S phase of the next cycle (Sanger et al., 1985). Cytokinesis altogether is absent during megakaryocyte differentiation, and can also be arrested at a late stage in spermatocytes of most species (Pepling et al., 1999; Vitrat et al., 1998). Cytokinesis starts by the specification of a cleavage plane and is followed by the ingression of an actomyosin contractile ring. During this transition, the mitotic spindle rearranges at the midzone to form an electron-dense structure known as the midbody, at the center of the intercellular bridge. Finally, by a process called abscission, daughter cells become physically separated. Although abscission failure can lead to tetraploidization, it remains poorly characterized due to several technical challenges (Steigemann and Gerlich, 2009). It is difficult to synchronize cells for the transient abscission process, which hampers most biochemical studies. Proteins involved at this stage may also be required earlier in the cell cycle and genetic mutations in the corresponding genes are thus likely to mask late functions. Finally, the midbody is beyond the resolution limit of most microscopes. Nevertheless, it has become clear that abscission requires remodeling of the membrane and cytoskeleton (Guizetti and Gerlich, 2010). Endosome delivery to the midbody and severing of the microtubule bundle are essential for correct abscission. The upstream regulators that initiate abscission remain unknown however. It is also unclear what regulates the timing of abscission, which can vary from minutes to hours, or can even remain incomplete, as in germ cells (Pepling et al., 1999). In our genetic screen for mutants affecting the early steps of oogenesis, we have identified the first mutations in the Drosophila homologues of Aurora-B and Survivin, two members of the Chromosome Passenger Complex (CPC). This complex regulates multiple steps of mitosis and is widely studied for its function at the centromeres, at the initiation of cytokinesis, and more recently during abscission. We found that Aurora B and Survivin inhibit abscission during normal development of Drosophila germ cells, using an allelic series of mutations. This inhibition is mediated by an Aurora B-dependent phosphorylation of Cyclin B, as a phosphomimic form of Cyclin B rescues premature abscission caused by a loss-of-function of Aurora B. In addition, we show that cell-type specific organelles, such as the fusome, and transcriptional programs, mediated by Bam in germ cells, impinge on this regulatory loop and can block abscission completely. We propose that the mutual inhibition between Aurora B and Cyclin B that we describe in germ cells, is likely to regulate abscission timing in a wide variety of cell types (Mathieu et al., Developmental Cell, 2013 Aug 12; 26(3):250-65) 2-2) Regulation of abscission downstream of AuroraB-CycB-Cdk1: the ESCRT-III complex AuroraB and Cdk1 do not perform the membrane scission event required for the final separation of daughter cells. The secondary constriction is thought to be formed, and then abscised, by the ESCRT machinery, and the vacuolar protein sorting 4 (VPS4) 14. The subunits of the ESCRT-III complex, CHMP4B and VPS4, are relocated at the exact site of the cut just before abscission occurs. A direct phosphorylation of the ESCRT- III CHMP4C by AuroraB has been documented recently in mammalian cells, and shown to be involved in the control of abscission timing 15-16. CHMP4C has an inhibitory effect on abscission, whereas CHMP4B, a translated splice variant, has a positive effect on abscission. We have shown that AuroraB positively regulates abscission, and CyclinB-Cdk1 inhibits it. It is tempting to speculate that AuroraB phosphorylates CHMP4C, and Cdk1 phosphorylates CHMP4B. However, there is only one homolog of CHMP4 in Drosophila called Shrub (or Shrb/SNF7) 17. We have recently shown that it acts downstream of AuroraB to regulate abscission in GSCs (Matias et al., 2015). Shrb mutants give rise to egg chambers with 32 cells due to the formation of stem cysts, confirming our previous model (Mathieu et al., 2013). Shrb/SNF7 seems, however, to have both a positive and negative effect on abscission. We showed that the ESCRT-III protein Shrb localizes to the midbody of the dividing GSC, functioning to promote abscission. Indeed, we found that reduced levels of Shrb resulted in the blockage, or strong delay, of abscission in the GSC and formation of a structure similar to a cyst. In these so called stem- cysts, the GSC keeps dividing while interconnected to its daughter cells. Consequently, we saw the appearance of egg chambers formed of 32 cells, instead of 16. Furthermore, Shrb function in abscission seems to be counteracted by AurB, as reducing AurB levels in Shrb heterozygous resulted in decreased stem-cysts and 32-cell cysts. Finally, Lethal giant discs (lgd), required for Shrb function in the endosomal pathway, was also seen localizing at the midbody and regulating abscission in GSCs. Removing one copy of Lgd from Shrb heterozygous increased the number of stem-cysts, but surprisingly the number of 32- cell cysts was reduced. This paradoxical result was explained with the observation of late abscission events in mitotic cysts, which divided the 32-cell cysts in the middle, leading to the formation of two cysts of 16 cells. (Matias et al., PLoS Genetics, 2015 Feb 3;11(2):e1004653) 2) How do homologue chromosomes find each other during meiosis? view more... We are studying the potential role of the cytoplasmic cytoskeleton in regulating chromosome organization during the early steps of meiosis in the germarium. Our lab has a longstanding interest in investigating the relationships between microtubules and the early stages of meiosis 18. How can this cytoplasmic cytoskeleton influence nuclear organization? A direct link between the cytoplasmic and nuclear sides remains mysterious. A first cue came very recently when we found that homologous chromosomes start to pair when reaching the nuclear envelope in the mitotic zone5. Once at the membrane, centromere movements are highly dynamic and we recently demonstrated that these movements are caused by rotations of the entire nucleus driven by microtubules and the SUN/KASH complex in females (Christophorou et al, Nature Cell Bio, 2015). Meiosis is a two-step cell division process (meiosis I and meiosis II) generating gametes. During meiosis I, each chromosome has to find its homologue in order to pair, synapse and initiate recombination and chromatin exchange between homologues. These events lead to the correct segregation of each homologue into separate daughter cells. Errors in any of these processes can result in aneuploidy, which leads to severe birth defects and miscarriages. In C. elegans, chromosome pairing depends on movements driven by cytoplasmic forces that act via members of the SUN and KASH domain proteins at the nuclear membrane (Penkner et al., 2009; Sato et al., 2009). Following homologue chromosomes pairing, synapsis takes place, a process that involves the formation of the synaptonemal complex (SC), a proteinaceous structure that forms between chromosomes and reinforces pairing. Drosophila is a powerful genetic system and the initiation of meiosis in germinal cells oogenesis is a useful model to study the mechanisms regulating homologue chromosome pairing and synapsis. In the germarium a series of mitotic divisions occur that give rise to a cyst of 16 cells interconnected by a cytoplasmic structure called the fusome (de Cuevas and Spradling, 1998). Among the cyst cells, one of them will differentiate as the oocyte while the rest of cells will adopt a nurse cell fate (Huynh and St Johnston, 2004). When the cyst has formed, meiosis, as seen by the formation of the SC is initiated in all 16 cells of the cyst (Carpenter, 1975; Huynh and St Johnston, 2000; King, 1970). Gradually meiosis becomes restricted to the future oocyte, while the 15 sister cells become nurse cells and lose the SC. We have recently shown that meiosis starts during the preceding mitoses in the fly germarium (Christophorou et al. PLoS Genetics, 2013)! We showed that in contrast to every cell types described so far in Drosophila, homologous chromosomes are not paired in germline stem cells. We further showed that during the differentiation of the daughter cell of the stem cell, homologous chromosomes become progressively paired. One surprising result is that this pairing occurs during the four mitosis preceding the entry into meiotic prophase. So, these mitoses “pre-pair” chromosomes for meiosis. In addition, we uncovered parts of the molecular mechanisms underlying this novel process by identifying two components of the synaptonemal complex (i.e. considered specific to meiosis) expressed during the four mitoses (Christophorou et al. PLoS Genetics, 2013). These two components (C(3)G/Zip1 and Corona) localize at the chromosome centromeres in the mitotic region, and homologues pairing is greatly affected in their absence. Our results thus demonstrate that there is an active and de novo pairing of homologous chromosomes before entry in meiosis. In germ cells, meiosis is thus intimately linked with mitosis. Our results challenge current models and should change people’s thinking of chromosome organization during entry in meiosis. Cell biologists tend to study cytoplasmic and nuclear organizations separately. However, increasing evidence suggests that cytoskeletal forces can directly influence chromosome organization within the nucleus. The pairing of meiotic chromosomes is a perfect case study to address this biological question. Indeed, at this stage, each chromosome needs to move within the nucleus to pair with their unique homologue, while the nuclear envelope is still intact. This question has been investigated in a few model organisms and has revealed a bewildering diversity of mechanisms such as actin-driven telomere movements in budding yeast, microtubule-driven horsetail motion in fission yeast, and very recently telomere rotations in mouse spermatocytes (Shibuya et al., 2014, Nat. Cell Bio). Surprisingly, this question remains completely unexplored in Drosophila, despite being a leading model system to study meiosis. One reason is that meiotic pairing was not known to even exist in Drosophila, as it was believed that homologous chromosomes were always paired in somatic and germline cells. We and others have shown very recently that instead chromosomes were not paired in germline stem cells, and required pairing before entering meiosis (Christophorou et al. PLoS Genetics, 2013 and see also Cahoon and Scott Hawley, PLoS Genetics, 2013). It opened a complete new field to understand the underlying cellular mechanisms. We have developed fast and ultra-fast live-imaging of these stages, based on techniques we had pioneered (Fichelson et al., 2009, Nat. Cell Bio), to investigate pairing in Drosophila. We found that meiotic nuclei performed cycles of complete rotations required for chromosome pairing. We were able to photo-tag with a laser any subpart of the nucleus and characterize quantitatively that nuclei were rotating as units, which is a novel phenomenon in meiosis to our knowledge. In addition, we have uncovered the molecular mechanisms required for nuclear looping and chromosome pairing. We demonstrated that: 1) the force-generators are the microtubules and the motor Dynein; 2) the force-transmitters at the nuclear envelope are the Drosophila SUN/KASH homologues, Klaroid and Klarsicht respectively. We showed that they link chromosome centromeres to the microtubule cytoskeleton; 3) we showed that the dynein- interacting protein Mud (NuMA in vertebrates) colocalizes with KASH and Dynein at the nuclear envelope and that Mud is required to maintain the integrity of the nuclear envelope and to assemble the synaptonemal complex. We thus identified a novel function for Mud/NuMA, which is to resist mechanical forces exerted on the nuclear envelope during nuclear rolling. Overall, our study reveals a novel cellular phenomenon, which is rolling nuclei in Drosophila, and uncovers a novel pathway (centromere/SUN/KASH/Mud/Dynein), which transmits and resists cytoplasmic forces during meiosis. (Christophorou et al. Nature Cell Biology, 2015)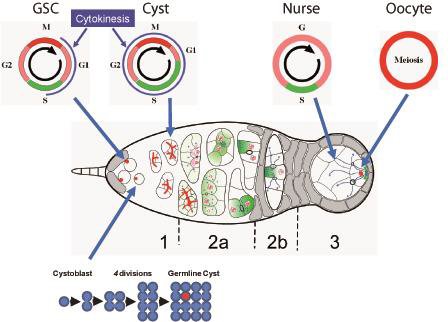
Our lab has pioneered the use of live imaging of the germarium, and has set up for the first time conditions allowing for the monitoring of single stem cells in their intact microenvironment. This allowed us to uncover the mechanisms underlying the asymmetric growth of GSCs and, more recently, to study the regulation of abscission duration in these cells.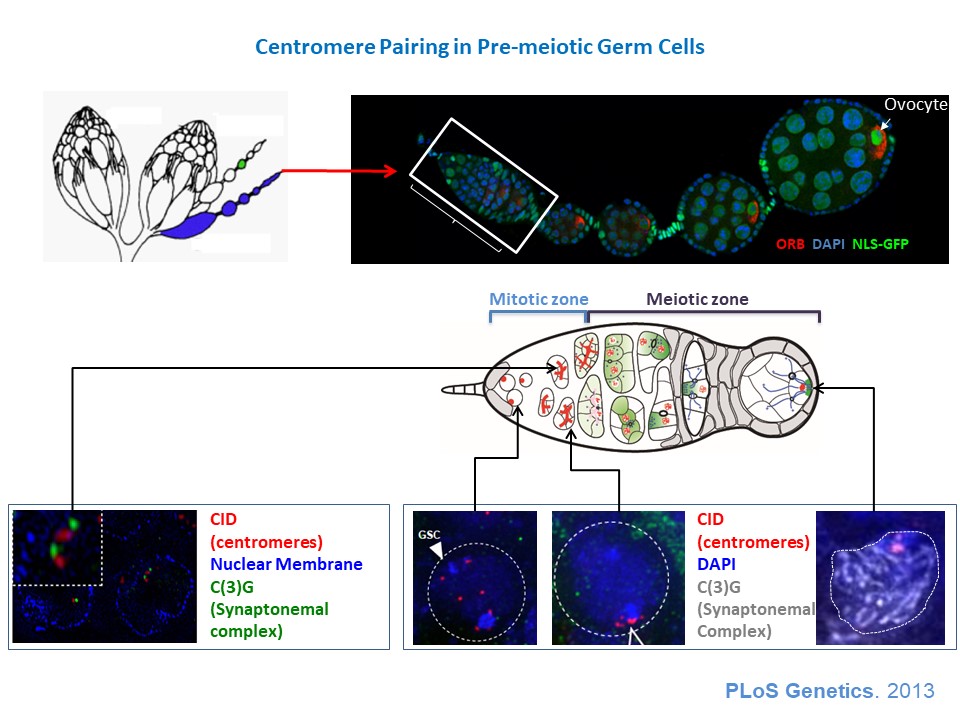
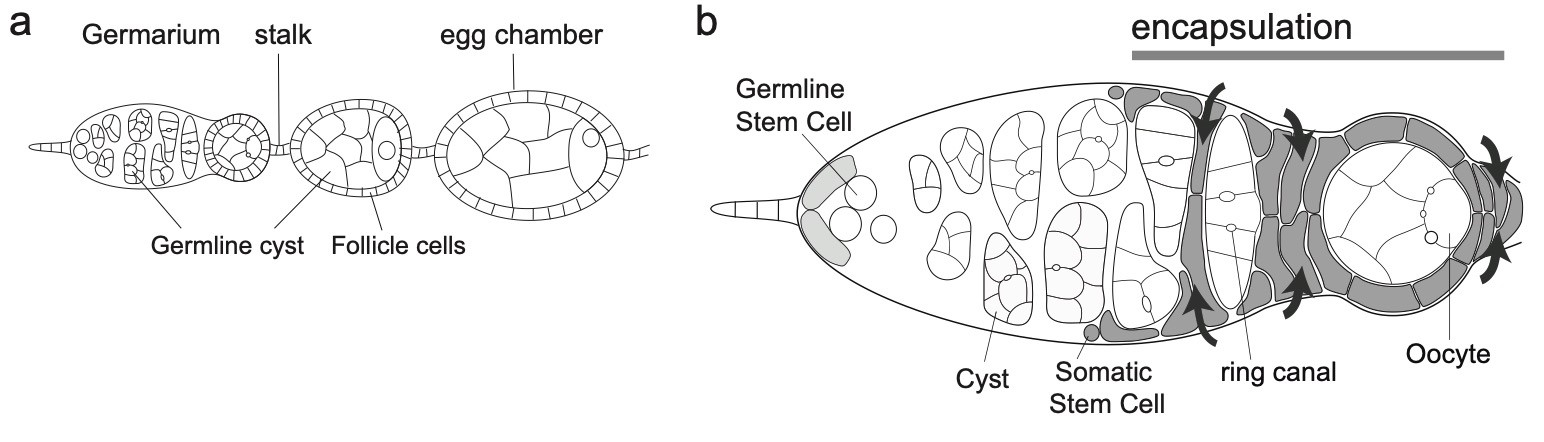
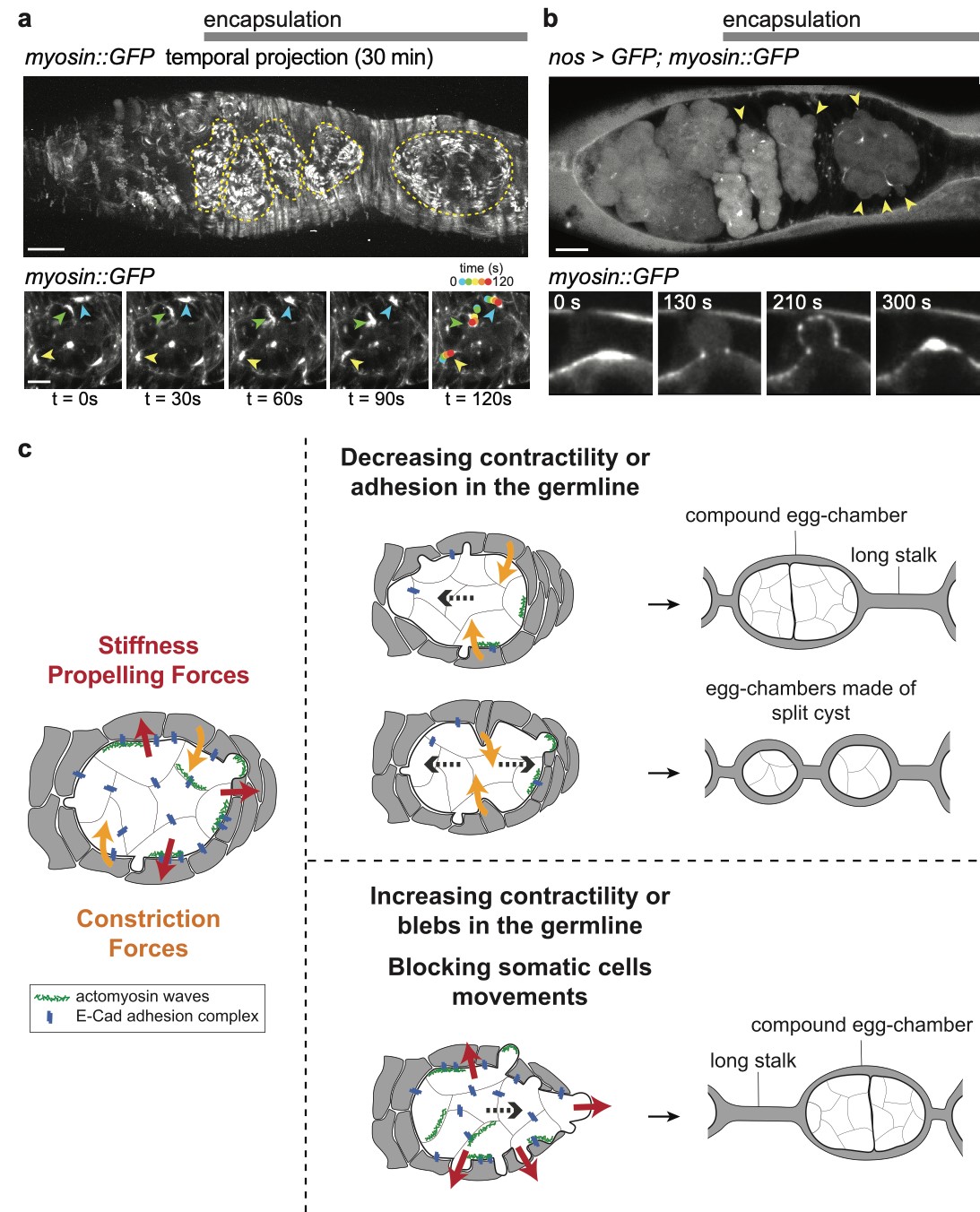
3) How is the germline genetic material protected from DNA damages? view more... The genetic information contained in the female gamete needs to be safely transmitted to the next generation. We are studying how mobile DNA elements such as transposons are silenced in germline cells. One mutation stood out in our screen because homozygous mutant flies were viable, but females were 100% sterile and males were sub-fertile (Jagut et al., 2013). Mutant ovaries are rudimentary and egg chambers do not develop past stage 6-7. This phenotype suggests a specific function for the mutated gene in germ cells development. To our surprise, the molecular identification of the mutation showed that the disrupted gene was rpp30, which encodes a regulatory subunit of the RNase P (Esakova and Krasilnikov, 2010). This nuclease performs an ubiquitous function in cleaving the 5’-end of pre-tRNAs in every cell types. Many defects could cause the early arrest in oogenesis, such as growth defects, polarity defects or DNA damages. We found that removing chk-2, the DNA-damage checkpoint in Drosophila, at the same time as rpp30, partially rescued oogenesis arrest, and even restored some fertility. This result shows that the main cause of arrest is the presence of DNA lesions in rpp30 mutant flies. It has been hypothesized that one source of DNA damages during early oogenesis is the activity of transposable elements in the genome. When these mobile DNA elements move in the genome, they can disrupt protein-coding genes, alter transcriptional regulation, and cause chromosomal breakage and large-scale genomic rearrangement (Aravin et al., 2007; Brennecke et al., 2007; Brennecke et al., 2008; Malone et al., 2009). In normal conditions, transposons activity is prevented by the presence of small RNAs associated with the Piwi protein (piRNAs), which recognize transposon sequences and cleave their precursors. We found by RT-qPCR that many transposons were highly transcribed in rpp30 mutant ovaries compared to wild type. Deep sequencing of mutant ovaries further revealed that piRNAs levels were greatly diminished. We found that in the double mutant rpp30, chk-2, piRNAs levels were restored to almost wild type levels, indicating that Rpp30 cannot be directly involved in processing piRNAs. Our genomewide RNAseq analysis of rpp30 mutant revealed that piRNA clusters transcription was specifically affected. At present, our favorite hypothesis to explain an indirect effect of tRNA biogenesis on piRNAs transcription is through the creation of replication stress between tRNA clusters and piRNA clusters. Indeed, we noticed that tDNA genes are not randomly localized within the genome but are clustered closed to piRNA clusters and other repeated sequences. Defects in tRNA co-transcriptional processing could impede replication of the nearby piRNAs clusters and subsequently impair their transcription. We have two arguments in favor of this model: 1) double mutant rpp30; claspin rescue oogenesis. claspin is a checkpoint protein specifically responding to replication stress (more specifically than chk-2); and 2) ChIP-qPCR experiments show that epigenetic marks were specifically absent from piRNAs clusters localized nearby tRNA clusters in rpp30 mutant ovaries. (Molla-Herman et al., The EMBO Journal, 2015). During the course of our analysis of small RNA sequences in wild type and rpp30 mutant ovaries, we identified a novel class of small RNAs derived from tRNA sequences in Drosophila. These fragments are not degradation products of full length tRNAs, as they have specific sizes and are derived from specific sequences at the 5’ and 3’ of pre-tRNAs. We are developing a bioinformatics pipeline to analyze this novel class of small RNAs in collaboration with the lab of Christophe Antoniewski (UPMC, Paris). These tRNA-derived RNA fragments (tRFs) have also been recently described in deep sequencing analysis of mouse and human cells, and thus exist in a wide range of organisms (Haussecker et al., 2010; Pederson, 2010). Their function remains however completely unknown. In addition to the bioinformatics part of the project, we are also developing tools to detect these tRFs and their activity in vivo with transgenic fluorescent sensors. These tRFs are particularly exciting because most of them contain sequences able to pair with retro-transposons sequences. Indeed, retro-transposons, like retro-viruses, use tRNAs as primer for retrotranscription. The 5’ or 3’ parts of tRNA can thus base pair with TE sequences. We are thus exploring the possibility that tRFs could guide Aubergine or Ago3 to TEs RNAs, and induce their cleavage. This novel class of small RNAs could be an additional source of protection of the genome against transposable elements. 4) How are the germline and somatic tissues coordinated for the morphogenesis of the egg chamber ? view more... During development, tissues and cells from different origins cooperate and coordinate their morphogenetic movements to shape embryos and generate complex organs. Proper coordination of cells and tissues behavior requires mechanical signals operating in tandem with biochemical cues (Chanet and Martin, 2014; Zhang and Labouesse, 2012). Oogenesis represents a conserved program involving two tissues, germline cells and supporting somatic cells that have to coordinate their growth, differentiation and morphogenesis to give rise to the future female gametes. Germ cells undergo several rounds of mitosis before entering meiosis. In most species, these mitoses are incomplete giving rise to cysts of cells interconnected by cytoplasmic bridges. Each germline cyst is then surrounded by cells of somatic origin called pre-granulosa cells in mammals and follicle cells (FCs) in Drosophila. In mammals, pre-granulosa cells invade in between germ cells and each cyst eventually breaks down into single cells encased by granulosa cells, forming primordial follicles. In Drosophila, FCs encapsulate cysts made of precisely 16 interconnected germ cells to form an egg chamber. How somatic and germ cells coordinate their movements and behaviors to form primordial follicle (mammals) and egg chambers (flies) is unknown. In Drosophila, the early steps of oogenesis take place in the germarium, a specialized structure that contains both germline and somatic stem cells (Huynh and Johnston, 2004). The germarium produce egg chambers throughout adult life. The egg chamber is made of 16 germline cells (15 nurse cells and one oocyte) surrounding by an epithelial layer of somatic FCs. It is the basic unit of female reproduction, that will grow and differentiate into a fertile egg. Germline stem cells (GSCs) are located at the most anterior tip of the germarium and generate cysts of 16 cells interconnected by ring canals. Once made of 16 cells, germline cysts come into contact with FCs produced by follicle stem cells (FSCs). Encapsulation starts when FCs migrate centripetally and separate germline cysts from each other. Then, FCs intercalate and form stalks of cells, which pinch off the newly formed egg chamber from the germarium, a process called egg-chamber budding (Morris and Spradling, 2011; Wu et al., 2008) (Figure 1). The mechanisms by which FC precursors precisely recognize and engulf an individual cyst are unknown. Figure 1: Egg-chamber formation (a) Scheme of an ovariole. The Drosophila ovary is composed of 16-20 ovarioles, each of which contains a chain of progressively more and more mature egg chambers. The egg chamber consists in 16 germ cells, surrounded by an epithelial layer of follicle cells. Egg chambers are produced throughout adult life by a structure called the germarium. (b) Close-up on a germarium. The germarium contains two populations of stem cells, germline stem cells that produce germ cells, and follicle stem cell, that produce somatic follicle cells (gray). In the anterior part of the germarium, germ cell progenitors divide but with incomplete cytokinesis to generate cysts of 16 cells interconnected by ring canals. Encapsulation occurs in the second half of the germarium, when germline cysts become surrounded by FCs that migrate centripetally around each cyst and intercalate (arrows) to progressively pinch of the newly formed egg chamber out of the germarium. We recently discovered that mechanical coordination between the germline and the somatic tissues is essential for proper encapsulation. We found that germline cells generate mechanical forces to maintain their integrity and position in the germarium during encapsulation, counterbalancing the forces exerted by ingressing FCs. Reducing contractile forces generation in germ cells or their transmission to the surrounding FCs lead to encapsulation defects (Chanet and Huynh, 2020). Mechanical forces are produced by cytoskeletal machines such as actomyosin meshworks that often display dynamic behaviors. To be effective, these mechanical forces must be transmitted to the extracellular substrate or neighboring cells through links between F-actin network and junctional complexes, such as cadherins and integrins (Lecuit et al., 2011; Salbreux et al., 2012). We observed that germline cysts generate cortical contraction waves at the time of their encapsulation by FCs (Figure 2a). This cortical contractility is associated with the formation of blebs that project into the somatic layer (Figure 2b). We found that reducing cortical contractility in germ cells leads to the formation of defective egg chambers containing too few or too many germ cells (Chanet and Huynh, 2020). Abnormal egg chambers containing defective number of germ cells are not fertile. Live imaging revealed that these defects arise in the germarium at the time of encapsulation. Germline cysts with reduced cortical contractility were pushed backward by constricting FCs, which can provoke collision with the next cyst. Such collision can lead to the formation of a compound egg chamber containing two cysts packaged together and a long stalk of somatic cells devoid of germ cells. We also observed germline cysts with reduced cortical contractility being squeezed and cut in two separate units by ingressing FCs. Such cyst splitting events give rise to egg chambers with too few germ cells (Figure 2c). Together these results indicate first that cortical contractility in germ cells is required to maintain cysts positioning and avoid collisions between cysts, which can result in the formation of compound egg chamber and long stalks of FCs devoid of germ cells; second, germ cells contractility is also required to maintain cyst integrity as a group of 16 cells and confer stiffness to avoid cyst splitting. Reducing E-Cad adhesion between germline cysts and FCs induce similar encapsulation defects indicating that transmission of contractile forces through E-Cad based junctions is required for correct encapsulation. Finally, we showed that either increasing germline cyst activity (by increasing contractility in germ cells or facilitating blebbing) or blocking constriction forces exerted on it by surrounding FCs (by mechanically blocking their movement around germline cyst) induces visible migration of the cyst toward the posterior of the germarium. Speeding up cyst migration can also lead to collision with the posterior cyst and formation of compound egg chambers and long stalks. We thus conclude that germline cysts generate stiffness and propelling forces that counterbalance somatic constriction forces and help maintaining cyst integrity and position at the time of encapsulation (Chanet and Huynh, 2020) (Figure 2c). Our study set up the bases for a mechanical understanding of encapsulation and revealed a prime role of the germline cysts in this process that were previously thought to be passive. Figure 2: Germline cysts produce mechanical forces required for their correct encapsulation (a) Cortical waves around germ cell cortexes. (top) Temporal projection of a 30 min movie of a germarium expressing sqh::GFP that stains myosin. Germline cysts (outlined with yellow dotted lines) generate contraction waves at the time of encapsulation. (bottom) Time-laps images of sqh::GFP signal showing myosin travelling waves around the cell cortexes of a germline cyst. On the last image, waves displacement is represented with time-colored dots. Scale bars, 10mm (top), 5mm (bottom). (b, top) cytoplasmic expression of GFP in the germline reveals the presence of bleb protrusions (arrows). (bottom) still-images following a bleb formation and retraction, sqh::GFP stains myosin. Scale bar, 10mm. (c) Encapsulation requires a proper balance between germline and somatic forces. (left) Germline cysts exert mechanical forces dependent on cortical contractility and adhesion (red arrows). This confers stiffness within the 16-cell cyst to maintain cyst integrity and propelling forces to maintain proper cyst positioning during encapsulation. At the same time, somatic cells migrate around germline cyst exerting constriction forces (orange arrows). In wild type, germline forces resist constriction forces exerted by surrounding somatic cells and maintain correct cyst position during encapsulation. (middle) Modifying the equilibrium between germline and somatic forces decouples cysts movement from somatic cells movement. On one hand, decreasing germline contractility or adhesion induce germline cysts sliding backward, leading to cysts collision, which can lead to the formation of compound egg chambers. Decreasing germline contractility or adhesion also induce cyst splitting by constricting somatic cells, which generate incomplete egg chambers. On another hand, increasing germline contractility or blebbing or blocking somatic cells movement can induce a net forward migration of the germline cysts that can also result in cyst collision and encapsulation defects.